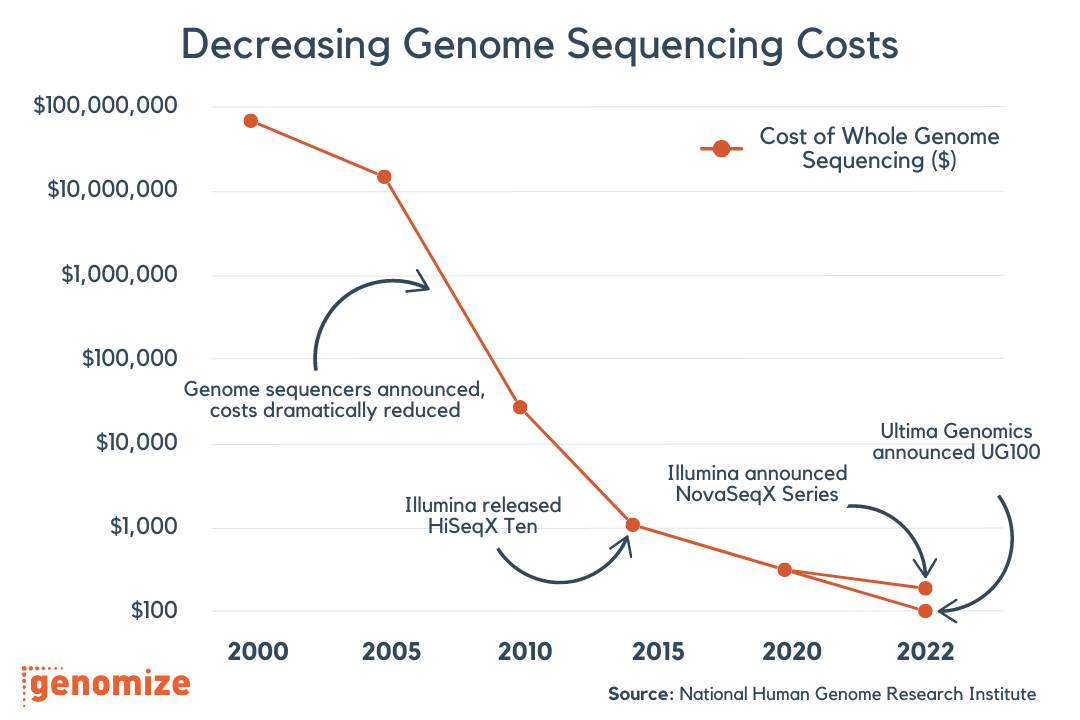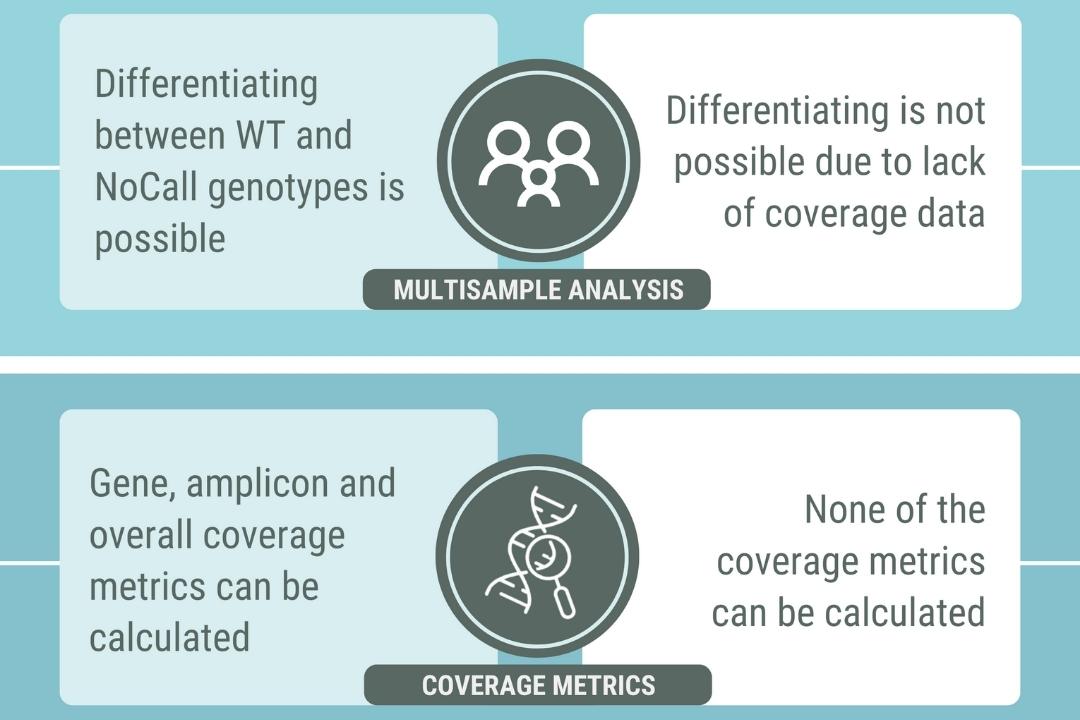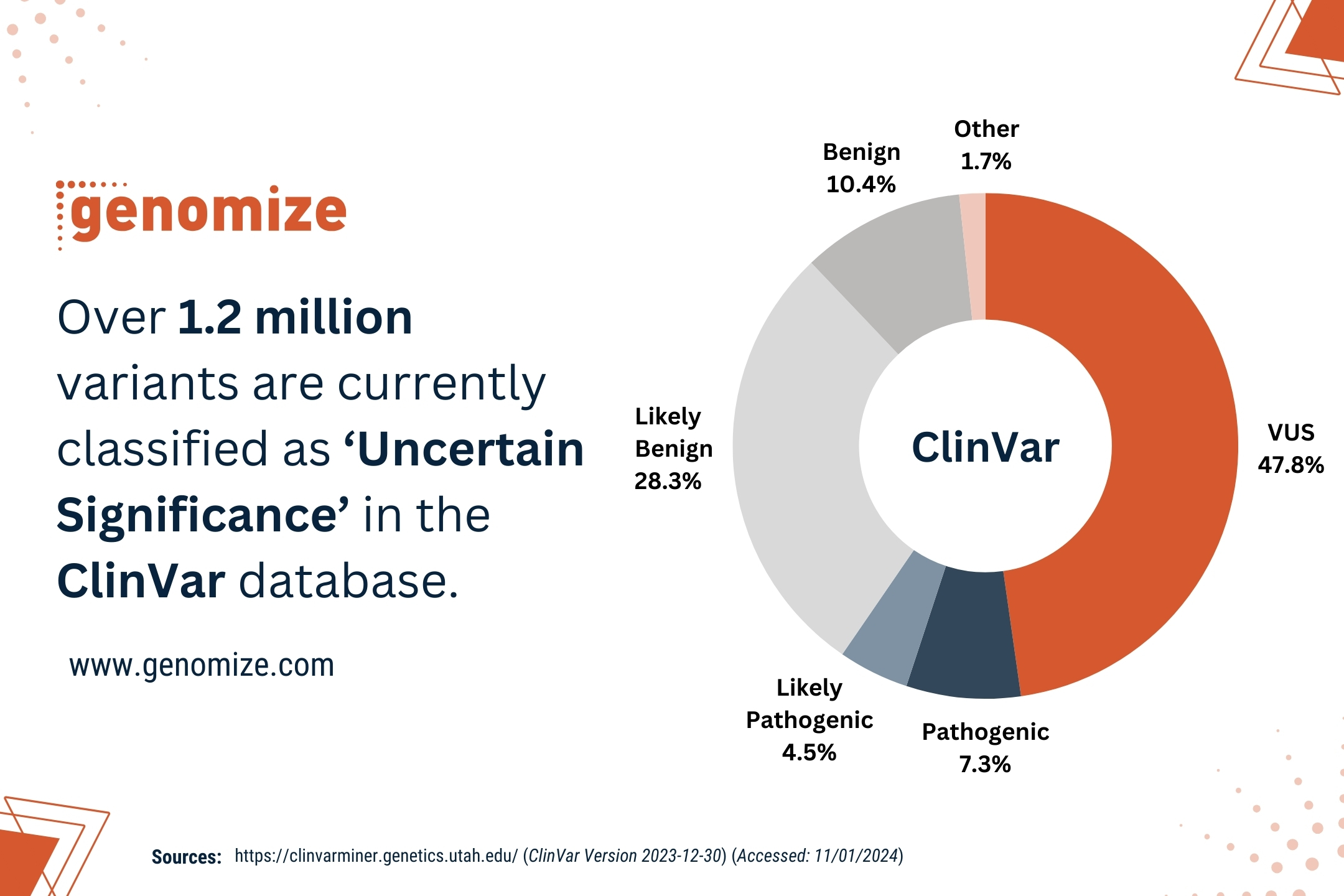



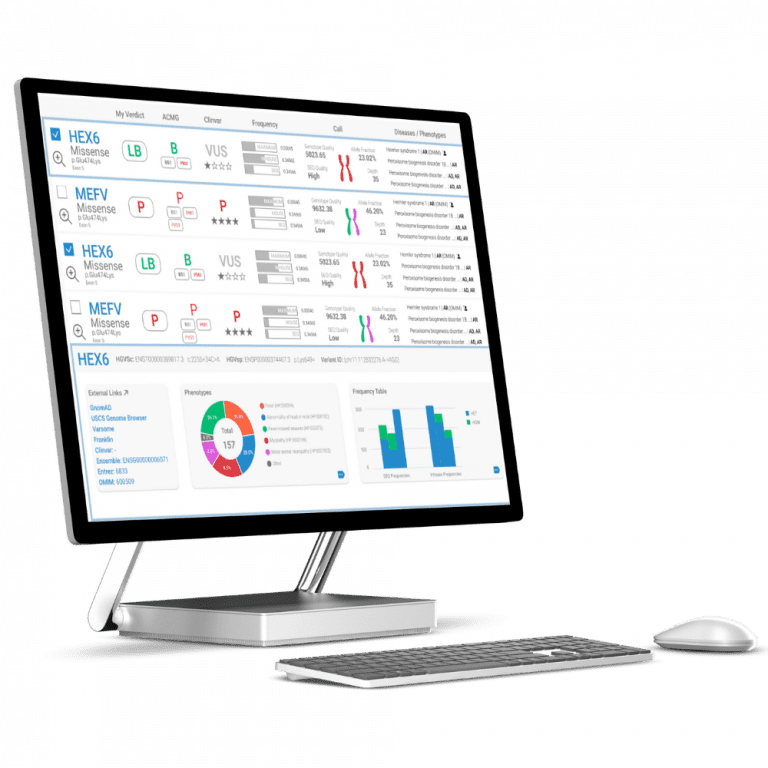

The SEQ Platform’s advanced bioinformatics pipelines and AI-driven variant prioritization enable accurate detection of genomic variants, leading to more precise diagnosis of genetic disorders.
Take control of your genomic data. With the SEQ Platform, you can easily build and manage your own genotype/phenotype database, empowering you to make informed decisions based on your unique genomic insights.





SEQ Platform’s highly sensitive somatic pipeline can help identify alterations and provide insights into potential treatment options based on ASCO/AMP/CAP guidelines.