
Short-read sequencing has transformed clinical genomics, with decreasing costs and faster turnaround making genome and exome level testing routine in rare disease diagnostics [1, 2]. Still, a substantial fraction of cases remain unsolved after standard short-read workflows. The reasons are well characterized: structural variants larger than a typical read may go undetected, repeat expansions may not be sized accurately, and variants in paralogous or repetitive regions may be lost when reads cannot be uniquely mapped [3, 4, 5].
Here, we describe how PacBio HiFi long-read sequencing addresses these specific blind spots, how we integrated HiFi-based analysis into the Genomize SEQ Platform, and how the resulting pipeline performs on the variant classes that account for most of the diagnostic gap.
HiFi long-reads in rare disease
The diagnostic gap traces back to a physical property: the read length. Short-read platforms typically generate reads around 150 bp, while PacBio HiFi reads span 15,000–25,000 bp [6, 7]. This difference has direct consequences for several variant classes.
In regions of high sequence similarity, including paralogs, pseudogenes, and segmental duplications, short reads frequently map to multiple locations with similar sequence, producing ambiguous alignment and misassigned variants. A single HiFi read can span an entire locus resolving the ambiguity directly [8]. Long reads also cover the full extent of most disease-associated repeats, allowing both length estimation and motif identification at loci such as HTT, FMR1, and RFC1 [9, 10]. Structural variants become detectable with higher reliability since long reads can capture the entire variant span rather than relying on breakpoint inference from split or discordant read pairs [11, 12]. Although short-read pipelines can be tuned to infer some variants in paralogs, repeats and large structural variants, long reads make these regions directly observable rather than inferred.
Phasing follows the same logic. Short-read workflows infer haplotype structure statistically or through trio-based analysis [13], whereas long reads carry phase information natively [14]. Because a single read can span tens of kilobases, two variants in the same gene can be assigned to the same allele (cis) or the other (trans), which is the determining factor for compound heterozygosity in recessive diseases. Trio data remains necessary to determine parental or maternal inheritance, but cis/trans status can be established from the proband alone.
Methylation is a separate gain in addition to the benefits of the read-length. HiFi reads carry native 5mC methylation tags, eliminating the need for a separate bisulfite library [15].
These capabilities are delivered by the analysis tools developed by PacBio, and they are now integrated into the SEQ Platform as described below.
Pipeline overview
We integrated the PacBio-recommended HiFi secondary analysis stack into the Genomize SEQ Platform (Table 1). Each component handles the variant class it was developed for, with two exceptions: within 12 medically relevant segmental duplication regions, including SMN1/SMN2, RCCX, STRC, NCF1, NEB, and GBA, Paraphase takes priority over DeepVariant, producing haplotype-aware variant calls and copy number determinations across paralogous gene families that confound standard alignment-based callers [18, 19]; and small variants, structural variants, and tandem repeat variants are jointly phased by HiPhase [14], producing phased genotypes that support compound heterozygous identification across small variants on opposite haplotypes.
Table 1: HiFi secondary analysis components integrated into Genomize SEQ.
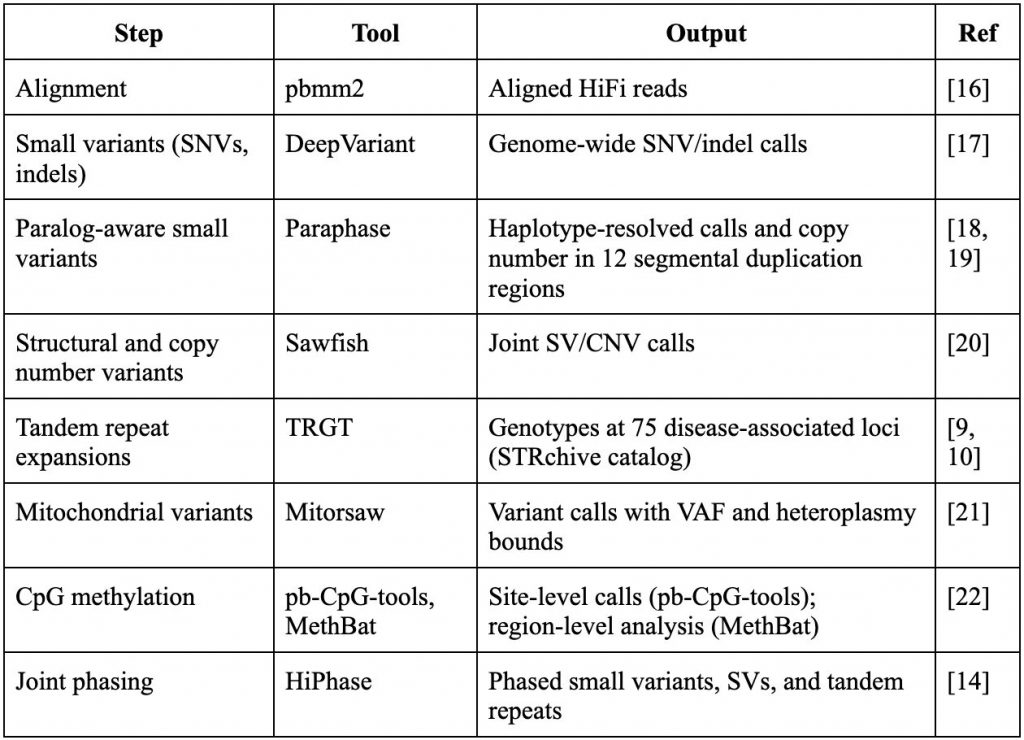
Listed tools reflect PacBio’s recommended HiFi secondary analysis stack at the time of publication and may change.
Evaluating the SEQ Platform using Genome in a Bottle truth sets
To evaluate our PacBio pipeline, we used HG002, a well-characterized GIAB reference sample. Variant calls were assessed against three GIAB truth sets covering progressively more of the genome’s difficult content: the v4.2.1 small variant benchmark [23], the Challenging Medically Relevant Genes (CMRG) benchmark covering 273 genes underrepresented in v4.2.1 [24], and the v5.0 benchmark derived from a diploid T2T assembly of HG002 [25]. For evaluations against v5.0, we used the GRCh38-mapped version to match the reference used by the pipeline.
Small variant accuracy
SNV F1 was near 1.00 across all three truth sets, with slightly lower indel F1 against the assembly-based benchmarks (Figure 1a). To evaluate variant calls at the haplotype level, we ran vcfdist against the two phased truth sets (CMRG v1.00 and v5.0 smvar); v4.2.1 is unphased and not evaluable with vcfdist. Unlike hap.py, vcfdist compares phased genotypes directly and awards partial credit when a variant is correctly identified but represented differently in the VCF [26, 27]. Phased F1 scores followed the same pattern as the unphased evaluation, with SNVs near 1.00 and modestly lower indel scores against both truth sets (Figure 1b). Indel F1 was higher against v4.2.1 than CMRG and v5.0 smvar in both evaluations, consistent with indels concentrating in homopolymer and tandem-repeat contexts that v4.2.1 underrepresents and the assembly-based truth sets cover more comprehensively.
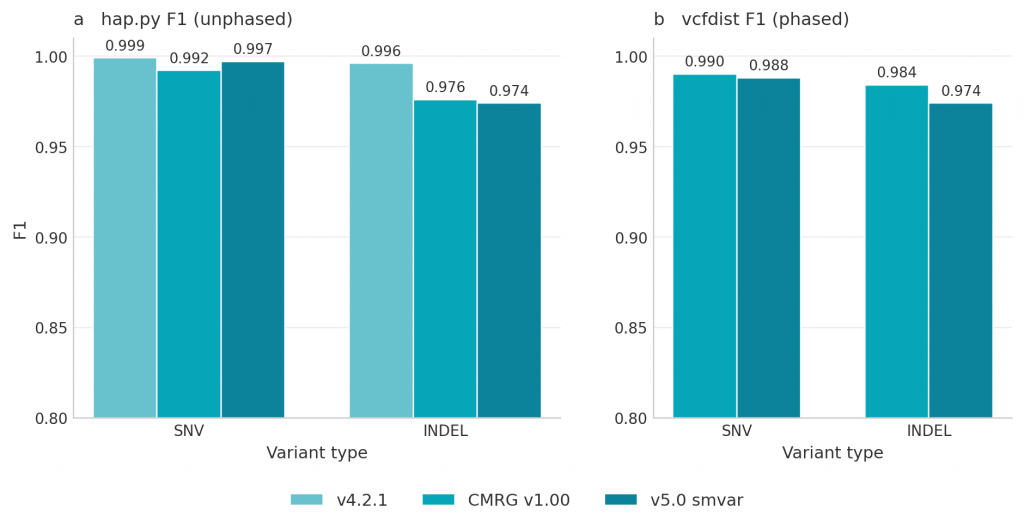
Figure 1: Small variant accuracy of the SEQ HiFi pipeline on HG002. F1 scores for the pipeline’s small variant calls, evaluated against three GIAB truth sets: v4.2.1, CMRG v1.00, and v5.0 smvar. (a) Unphased F1 (hap.py) for SNVs and indels against all three truth sets. (b) Phased F1 (vcfdist) for SNVs and indels against CMRG v1.00 and v5.0 smvar; v4.2.1 is not shown in panel b because it is unphased and not evaluable with vcfdist.

Figure 2: Structural variant accuracy of the SEQ HiFi pipeline on HG002. Truvari F1 scores for structural variant calls from the pipeline, evaluated against the CMRG-SV v1.00 and GIAB v5.0 stvar truth sets.
Structural variants present a known challenge for short-read sequencing because the read length constrains the ability to span breakpoints and resolve complex events [11, 12]. Long-read sequencing extends SV detection into regions short-read pipelines cannot reliably access [28].
SV F1 was 0.993 on CMRG-SV and 0.971 on v5.0 stvar (Figure 2). SV F1 was higher against CMRG-SV than against v5.0 stvar, reflecting CMRG-SV’s narrower focus on medically relevant genes compared with v5.0’s broader coverage into repeat and segmental duplication regions (Figure 2). Evaluation was performed against the GRCh38-mapped version of v5.0 stvar to match the pipeline’s reference coordinates.
Haplotype-resolved analysis: paralogs and compound heterozygosity
Long-read sequencing produces individual reads long enough to uniquely characterize the allele they reside, allowing variants to be assigned to haplotypes without statistical inference or trio sequencing. This native haplotype resolution underlies two clinically important applications: variant calling in paralogous gene families, and direct determination of compound heterozygosity from a single proband.
Standard short-read pipelines are developed for unique, single-copy genomic regions and often fail in paralogous regions [8]. When a short read aligns equally well to multiple paralogs, the alignment is assigned a low mapping quality score and is typically excluded, causing data loss across the paralog family. Clinically relevant examples include the RCCX module, SMN1/SMN2, and NEB, which are described in detail below.
When variants are called, they can be misassigned to the wrong paralog copy, misclassified as heterozygous, or merged across copies into incorrect copy-number calls [8, 29]. Many clinically important genes sit in these regions, including several with well-documented short-read diagnostic blind spots. Three examples below illustrate the range. Paraphase handles these regions by extracting all reads aligning to a paralog family, realigning them to a single archetype gene, phasing them into haplotypes, and calling variants and per-haplotype copy number on each resolved haplotype separately [18, 19]. Both outputs are shown in the SEQ targeted caller view.

Figure 3: Structure of RCCX module at chr6p21.3. Three pairs of paralogs are arranged as two tandem copies (RCCX module 1 and module 2; brackets). Arrows indicate gene orientation along the + and − strands. Solid outlines mark functional genes; dashed outlines mark pseudogenes. Teal fill highlights CYP21A2, the disease-relevant gene. Coordinates in Mb (GRCh38); the break on the right compresses the distance to TNXB.
RCCX on chromosome 6p21.3 is a 30-kb tandem module containing three pairs of paralogs (CYP21A2/CYP21A1P, C4A/C4B, TNXB/TNXA) and is highly variable in copy number across populations (Figure 3). 21-hydroxylase deficiency congenital adrenal hyperplasia arises from point mutations in CYP21A2, gene-conversion events transferring pseudogene sequence into the active gene, CYP21A1P–TNXB fusion deletions, and module copy-number changes [30, 31, 32]. Paraphase resolves the module into per-haplotype copies, reporting both variant calls and the number of RCCX modules on each allele.

Figure 4: SMN1, SMN2, and the SMN∆7–8 deletion haplotype at chr5q13.2. Exons are numbered along each gene. The red marker on SMN2 exon 7 indicates c.840C>T, the variant that distinguishes SMN1 from SMN2. On the SMNΔ7–8 track, exons 7 and 8 in red are absent in the deletion. Coordinates are shown in SMN1’s frame; SMN2 is drawn aligned to SMN1 rather than at its native coordinates (chr5:70.050-70.078 Mb).
SMN1 and SMN2 differ at only a small number of paralogous sequence variants. Among them, c.840C>T in exon 7 is the position that Paraphase and standard SMA diagnostics use to distinguish SMN1 from SMN2 (Figure 4). The substitution disrupts exon 7 splicing in SMN2 and is the basis for the clinical assay [19, 33]. Spinal muscular atrophy diagnosis depends on resolving this position and on quantifying SMN1 and SMN2 copy number as these characteristics modify disease severity and change the therapeutic response. Paraphase reports SMN1 and SMN2 copy number separately, along with the SMN∆7–8 deletion haplotype where present.

Figure 5: NEB locus at chr2q23.3. Top: the whole NEB gene with all exons shown as tick marks. Bottom: the triplicate region (TRI), made up of three tandem copies of the same eight-exon block (TRI-A, exons 82–89; TRI-B, exons 90–97; TRI-C, exons 98–105). NEB is on the − strand and is shown 5’→3′.
NEB contains a 32-kb triplicate region (TRI) made up of three nearly identical exon blocks (Figure 5). The normal copy number is six (three per allele), and one-copy gains or losses appear to be benign, while gains of two or more copies are associated with nemaline myopathy [34]. The TRI is collapsed by standard short-read alignment, which has historically prevented detection of these copy-number changes from sequencing data. Paraphase resolves the three TRI copies into separate haplotypes and reports per-haplotype copy number, which is the clinically actionable readout at this locus.
To assess Paraphase’s per-locus accuracy across this class of regions, we evaluated hap.py F1 on HG002 (DeepVariant 1.9 + Paraphase) at six clinically relevant paralog-family genes against the three GIAB truth sets (Figure 6): GBA (Gaucher disease and a Parkinson’s disease risk locus; near-identical GBAP1 pseudogene), NCF1 (autosomal recessive chronic granulomatous disease; pseudogenes NCF1B and NCF1C), PMS2 (Lynch syndrome; PMS2CL pseudogene with near-identical sequence over the 3′ end), SMN1 (described above), RCCX (described above), and STRC (autosomal recessive hearing loss DFNB16; STRCP1 pseudogene at ~99% identity).

Figure 6: Per-gene small variant accuracy of the SEQ HiFi pipeline across six clinically relevant paralog-family genes on HG002. hap.py F1 scores for variants called by DeepVariant 1.9 and Paraphase, evaluated against the v4.2.1, CMRG v1.00, and v5.0 smvar truth sets. Bars are shown only for truth set / gene combinations covered by the corresponding GIAB benchmark.
F1 ranges from 0.90 to 1.00 across loci, with v4.2.1 ≥ 0.95 at every gene. The harder truth sets show comparable performance with one exception: NCF1 drops to 0.90 on v5.0 smvar while remaining at 1.00 on v4.2.1, indicating that v5.0 smvar exposes calling difficulty in NCF1 regions that v4.2.1 does not capture (Figure 6).
Not every gene is evaluable against every truth set, because each truth set covers a different portion of the genome at varying confidence: PMS2 has a benchmark in v4.2.1 but is not represented in CMRG v1.00 or v5.0 smvar; RCCX, STRC, and GBA are evaluable in v5.0 smvar and v4.2.1 but not in CMRG v1.00; SMN1 is evaluable in CMRG v1.00 and v4.2.1 but not in v5.0 smvar. NEB is not represented in this evaluation because the pipeline and the GIAB truth sets define variant coordinates differently in the triplicate repeat region, preventing direct F1 comparison. The truth-set assignments shown reflect availability and coordinate compatibility rather than selective reporting.
A second application of native haplotype resolution is the direct determination of compound heterozygosity. In recessive disease, two variants in the same gene cause clinical phenotype only when they sit on opposite chromosome copies (in trans): two variants on the same copy (in cis) leave the other copy functional and are not disease-causing.
Short-read pipelines cannot distinguish these configurations from a single proband: when two variants are too far apart for any read to span both, the alignment alone cannot establish their phase. Trio sequencing or statistical phasing is required to assign variants to haplotypes, which adds cost and turnaround time.
A single HiFi read spans 15–25 kb, so two variants within that distance can be observed on the same read and assigned to the same haplotype directly. For variants separated by more than a read length, HiPhase [14] extends phasing by linking overlapping reads through shared heterozygous positions, producing phased genotypes from the proband alone. Trio data remains useful for assigning each haplotype to its parental origin, but cis/trans status, the determining factor for compound heterozygous diagnosis, can typically be established without it.

Figure 7: Direct determination of compound heterozygosity from phased HiFi reads.
Figure 7 illustrates the principle: two variants observed in a gene appear ambiguous under unphased calling (panel a), but resolve to opposite haplotypes once phasing is applied (panel b), establishing the compound heterozygous configuration.
Conclusion
We integrated the PacBio HiFi secondary analysis stack into the Genomize SEQ Platform and evaluated the resulting pipeline on HG002 against GIAB v4.2.1, CMRG v1.00, and v5.0. SNV accuracy was at or near 1.00 across all three truth sets; indel accuracy held above 0.97 even on the harder, assembly-based truth sets; structural variant F1 reached 0.986 on CMRG-SV and 0.950 on v5.0 stvar. Across six clinically relevant paralog-family genes, per-locus F1 ranged from 0.90 to 1.00.
Paraphase is the component that drives the most distinctive gain. By extracting reads from a paralog family, realigning them to a single archetype, and resolving haplotypes before variant calling, it converts regions that are diagnostic blind spots under standard alignment into regions where variants are called and assigned to the correct paralog copy. The same native haplotype information also supports compound heterozygous determination from a single proband, removing a workflow dependency on trio sequencing for cis/trans resolution.
The pipeline as integrated is suitable for use in research workflows on the variant classes that drive most of the short-read diagnostic gap such as structural variants, variants in paralogous regions, and tandem repeat expansions, while maintaining genome-wide SNV and indel accuracy comparable to established workflows.
References
1. Ewans LJ, Minoche AE, Schofield D, et al. Whole exome and genome sequencing in mendelian disorders: a diagnostic and health economic analysis. Eur J Hum Genet. 2022;30(10):1121–31. doi:10.1038/s41431-022-01162-2.
2. Faergeman SL, Andreasen L, Becher N, et al. Short-read genome sequencing at population scale: diagnostic insights from 2317 patients. Eur J Hum Genet. 2026. doi:10.1038/s41431-026-02089-8.
3. Eisfeldt J, Ek M, Nordenskjöld M, Lindstrand A. Toward clinical long-read genome sequencing for rare diseases. Nat Genet. 2025;57(6):1334–43. doi:10.1038/s41588-025-02160-y.
4. The 100,000 Genomes Project Pilot Investigators. 100,000 Genomes pilot on rare-disease diagnosis in health care — preliminary report. N Engl J Med. 2021;385(19):1868–80. doi:10.1056/NEJMoa2035790.
5. Stranneheim H, Lagerstedt-Robinson K, Magnusson M, Kvarnung M, Nilsson D, Lesko N, et al. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021;13(1):40. doi:10.1186/s13073-021-00855-5.
6. Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 2019;37(10):1155–62. doi:10.1038/s41587-019-0217-9.
7. Pacific Biosciences. How HiFi sequencing works [Internet]. [cited 2026 May 14]. Available from: https://www.pacb.com/technology/hifi-sequencing/how-it-works/
8. Kim SK, Jang J, Kim Y, et al. Integrative analysis of in silico predictions and clinical evidence to delineate the capability of HiFi long-read sequencing in paralogous genes. NPJ Genom Med. 2026;11:21. doi:10.1038/s41525-026-00555-2.
9. Dolzhenko E, English A, Dashnow H, De Sena Brandine G, Mokveld T, Rowell WJ, et al. Characterization and visualization of tandem repeats at genome scale. 2024.
10. Hiatt L, Weisburd B, Dolzhenko E, Rubinetti V, Avvaru AK, VanNoy GE, et al. STRchive: a dynamic resource detailing population-level and locus-specific insights at tandem repeat disease loci. Genome Med. 2025. doi:10.1186/s13073-025-01454-4.
11. Sedlazeck FJ, Lee H, Darby CA, Schatz MC. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat Rev Genet. 2018;19(6):329–346. doi:10.1038/nrg.2018.4.
12. Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun. 2019;10:1784. doi:10.1038/s41467-018-08148-z.
13. Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat Rev Genet. 2011;12(10):703–714. doi:10.1038/nrg3054.
14. Holt JM, Saunders CT, Rowell WJ, Kronenberg Z, Wenger AM, Eberle M. HiPhase: jointly phasing small, structural, and tandem repeat variants from HiFi sequencing. Bioinformatics. 2024;40(2):btae042. doi:10.1093/bioinformatics/btae042.
15. Tse OYO, Jiang P, Cheng SH, Peng W, Shang H, Wong J, et al. Genome-wide detection of cytosine methylation by single molecule real-time sequencing. Proc Natl Acad Sci U S A. 2021;118(5):e2019768118. doi:10.1073/pnas.2019768118.
16. Pacific Biosciences. pbmm2: a minimap2 frontend for PacBio native data formats [Internet]. GitHub. [cited 2026 May 8]. Available from: https://github.com/PacificBiosciences/pbmm2
17. Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36(10):983–7. doi:10.1038/nbt.4235.
18. Chen X, Baker D, Dolzhenko E, et al. Genome-wide profiling of highly similar paralogous genes using HiFi sequencing. Nat Commun. 2025. doi:10.1038/s41467-025-57505-2.
19. Chen X, Harting J, Farrow E, et al. Comprehensive SMN1 and SMN2 profiling for spinal muscular atrophy analysis using long-read PacBio HiFi sequencing. Am J Hum Genet. 2023. doi:10.1016/j.ajhg.2023.01.001.
20. Saunders CT, Holt JM, Baker DN, Lake JA, Belyeu JR, Kronenberg Z, et al. Sawfish: improving long-read structural variant discovery and genotyping with local haplotype modeling. Bioinformatics. 2025.
21. Pacific Biosciences. mitorsaw [Internet]. GitHub. [cited 2026 May 8]. Available from: https://github.com/PacificBiosciences/mitorsaw
22. Pacific Biosciences. MethBat [Internet]. GitHub. [cited 2026 May 8]. Available from: https://github.com/PacificBiosciences/MethBat
23. Wagner J, Olson ND, Harris L, Khan Z, Farek J, Mahmoud M, et al. Benchmarking challenging small variants with linked and long reads. Cell Genom. 2022;2(5):100128. doi:10.1016/j.xgen.2022.100128.
24. Wagner J, Olson ND, Harris L, McDaniel J, Cheng H, Fungtammasan A, et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat Biotechnol. 2022;40(5):672–80. doi:10.1038/s41587-021-01158-1.
25. Hansen NF, Dwarshuis N, Ji HJ, Rhie A, Loucks H, Logsdon GA, et al. A complete diploid human genome benchmark for personalized genomics. bioRxiv [Preprint]. 2025 Sep 21:2025.09.21.677443. doi:10.1101/2025.09.21.677443.
26. Cleary JG, Braithwaite R, Gaastra K, Hilbush BS, Inglis S, Irvine SA, et al. Comparing variant call files for performance benchmarking of next-generation sequencing variant calling pipelines. bioRxiv [Preprint]. 2015 Aug 2:023754. doi:10.1101/023754.
27. Dunn T, Narayanasamy S. vcfdist: accurately benchmarking phased small variant calls in human genomes. Nat Commun. 2023;14:8149. doi:10.1038/s41467-023-43876-x.
28. Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, et al. Characterizing the major structural variant alleles of the human genome. Cell. 2019;176(3):663–675.e19. doi:10.1016/j.cell.2018.12.019.
29. Ebert P, Audano PA, Zhu Q, Rodriguez-Martin B, Porubsky D, Bonder MJ, et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021;372(6537):eabf7117. doi:10.1126/science.abf7117.
30. Kim S, Yoon JH, Kim D, Park S, Kim GH, Yoo HW, et al. High-fidelity long-read sequencing reveals a complex RCCX locus at the single-nucleotide level in Korean patients with congenital adrenal hyperplasia. medRxiv [Preprint]. 2025 Jul 9:2025.07.09.25331238. doi:10.1101/2025.07.09.25331238.
31. Carrozza C, Foca L, De Paolis E, Concolino P. Genes and pseudogenes: complexity of the RCCX locus and disease. Front Endocrinol (Lausanne). 2021;12:709758. doi:10.3389/fendo.2021.709758.
32. Claps A, Kolomenski JE, Fernández F, Macchiaroli N, Ingravidi ML, Delea M, et al. High precision characterization of RCCX rearrangements in a 21-hydroxylase deficiency Latin American cohort using Oxford Nanopore long read sequencing. Sci Rep. 2025;15(1):24983. doi:10.1038/s41598-025-03799-7.
33. Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96(11):6307–6311.
34. Kiiski K, Lehtokari V-L, Vihola A, et al. A recurrent copy number variation of the NEB triplicate region: only revealed by the targeted nemaline myopathy CGH array. Eur J Hum Genet. 2016;24(4):574–580. doi:10.1038/ejhg.2015.166.



