

Alternative splicing allows a single gene to produce multiple protein isoforms with distinct structures and functions (Baralle & Giudice, 2017; Black, 2003) by joining different combinations of exons from a pre-mRNA to produce multiple different mature mRNA transcripts (Baralle & Giudice, 2017; Black, 2003). Alternative splicing is an essential mechanism for increasing the diversity of the proteome, and it plays a critical role in a wide range of biological processes, including development, differentiation, and disease (Baralle & Giudice, 2017; Black, 2003).
Around 98% of all human multi-exon genes are thought to undergo alternative splicing (Wang et al., 2008). Taking into consideration that 94% of all human genes are multi-exonic (Wang et al., 2008), it becomes clear that alternative splicing is heavily utilized in humans. It is not surprising that dysregulation of alternative splicing has been linked to a variety of human diseases, including cancer, neurological disorders, and genetic diseases (Baralle & Giudice, 2017).
In this article, we will approach another aspect of alternative splicing with direct implications for NGS data analysis and variant annotation. Although alternative splicing is essential for eukaryotes, it brings a crucial question to variant interpretation:
“Which isoform(s) should I interpret?”
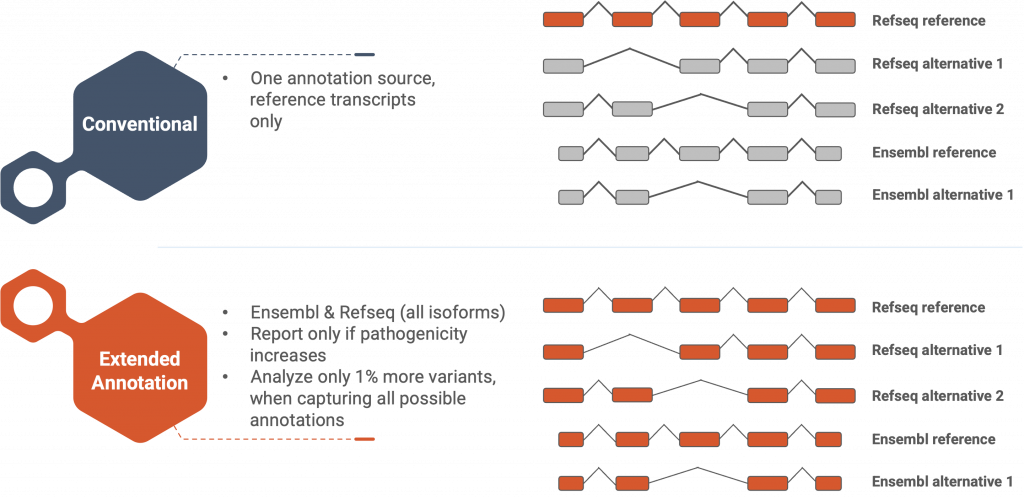
Figure 1: SEQ Platform’s Extended Annotation feature provides pathogenicity information for all alternative isoforms in both RefSeq and Ensembl databases.
The ACMG/AMP standards and guidelines for the interpretation of sequence variants recommend assessing clinically relevant isoforms, in addition to the reference transcript (Richards et al., 2015). However, variations in selecting relevant transcripts between laboratories can lead to the misinterpretation of variants, as highlighted by Schoch et al. (2020). Failure to consider alternative splicing and the most biologically relevant transcript has resulted in patients being undiagnosed or misdiagnosed (Schoch et al., 2020).
In addition to transcript selection, the choice of gene annotation source is another critical factor that can impact variant annotation and clinical diagnosis. The choice between the two most commonly used gene annotation sources Ensembl (Cunningham et al., 2022) and RefSeq (O’Leary et al., 2016) can and will affect the variant annotation due to discrepancies between the two highly trusted and acclaimed databases. Although the two databases share 27,031 genes, 33,732 gene annotations in Ensembl and 12695 gene annotations in RefSeq are unique (Chisanga et al., 2022). Even common genes in both databases can be annotated differently, leading to significant effects on data analysis. The MANE collaboration aims to consolidate and converge RefSeq and Ensembl gene and transcript annotations to address these issues. (Morales et al., 2022).
These factors lay a legion of extra burden on the genome analyst’s shoulders due to the sheer amount of manual labor it requires, and it is the NGS data analysis software’s duty to take over this burden and produce thorough and reliable results for the analyst. To address this issue, we have developed our “Extended Annotation” feature and it has been in our users’ service since early 2021.
Using the SEQ Platform, every variant is annotated for every transcript in both Ensembl and RefSeq databases, and their pathogenicities are calculated according to the ACMG guideline and consequent updates (Abou Tayoun et al., 2018; Richards et al., 2015). While Extended Annotation only increases the total number of annotations by 1% in an average WES sample, critical and clinically relevant changes in pathogenicity such as LB-to-VUS or VUS-to-LP (Figure 2) brought out to the analyst’s attention can affect diagnosis and treatment. The consequence changes in VUS-to-LP variants clearly show that the main contributors to this important change in pathogenicity are mainly due to the inclusion of “non-exonic” regions in the reference transcript in the CDS of alternative isoforms (Figure 3).
Our Solution
Using the SEQ Platform, every variant is annotated for every transcript in both Ensembl and RefSeq databases, and their pathogenicities are calculated according to the ACMG guideline and consequent updates (Abou Tayoun et al., 2018; Richards et al., 2015). While Extended Annotation only increases the total number of annotations by 1% in an average WES sample, critical and clinically relevant changes in pathogenicity such as LB-to-VUS or VUS-to-LP (Figure 2) brought out to the analyst’s attention can affect diagnosis and treatment. The consequence changes in VUS-to-LP variants clearly show that the main contributors to this important change in pathogenicity are mainly due to the inclusion of “non-exonic” regions in the reference transcript in the CDS of alternative isoforms (Figure 3).
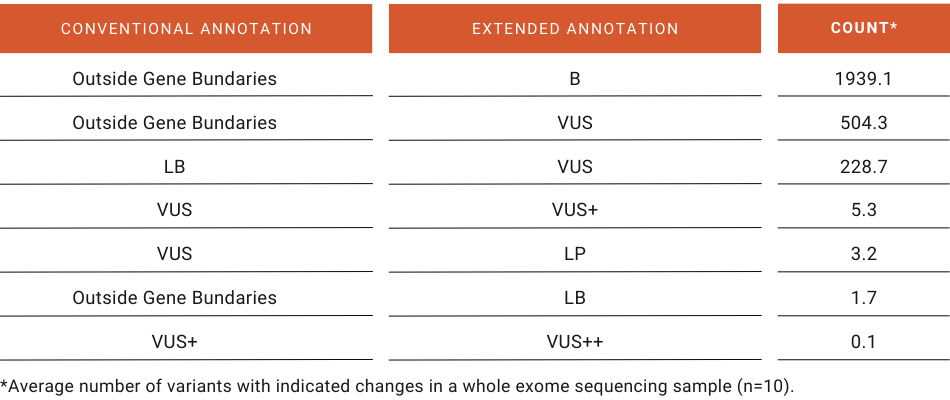
Figure 2: Top shifts in pathogenicity due to Extended Annotation feature in a whole exome sequencing sample.
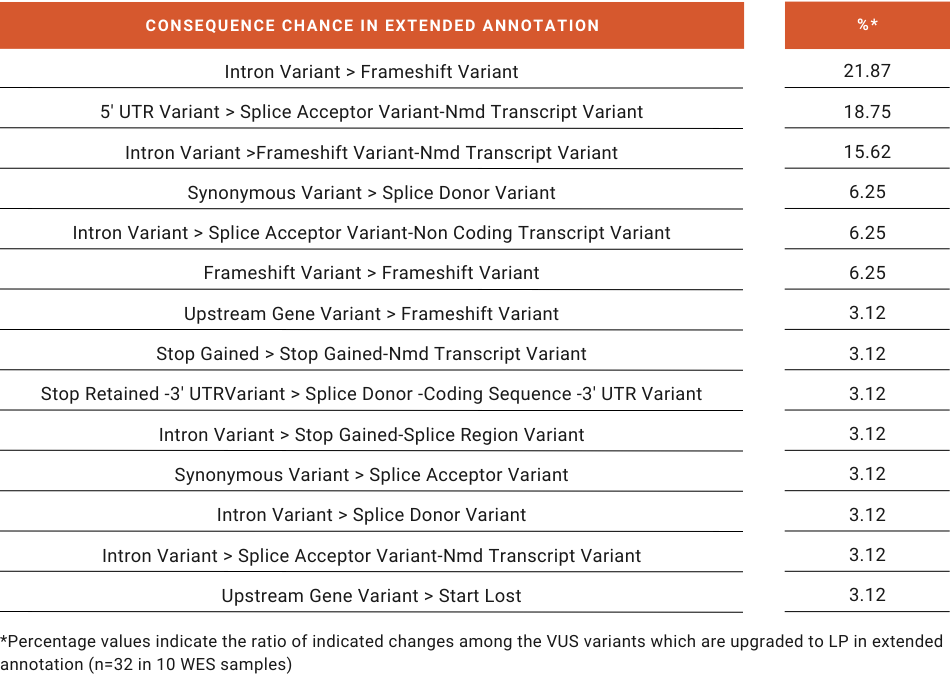
Figure 3: Consequence changes in VUS variants which are upgraded to LP in Extended Annotation.
With our unique design, the Extended Annotation feature exhausts all available information on the variant’s position without overwhelming the analyst with multiple annotations that do not contribute additional information for analysis. This way, analysts can quickly identify and prioritize clinically relevant changes, streamlining the variant interpretation process and improving the efficiency and accuracy of NGS data analysis.
With the Extended Annotation feature of the SEQ Platform, you can be sure that the ACMG pathogenicity classification for variants is correct, and that all transcripts and genes spanning the variant’s position are annotated.
References
Abou Tayoun, A. N., Pesaran, T., DiStefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., & Harrison, S. M. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Human Mutation, 39(11), 1517–1524. https://doi.org/10.1002/humu.23626
Baralle, F. E., & Giudice, J. (2017). Alternative splicing as a regulator of development and tissue identity. Nature Reviews Molecular Cell Biology, 18(7), 437–451. https://doi.org/10.1038/nrm.2017.27
Black, D. L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annual Review of Biochemistry, 72, 291–336. https://doi.org/10.1146/annurev.biochem.72.121801.161720
Chisanga, D., Liao, Y., & Shi, W. (2022). Impact of gene annotation choice on the quantification of RNA-seq data. BMC Bioinformatics, 23(1), 107. https://doi.org/10.1186/s12859-022-04644-8
Cunningham, F., Allen, J. E., Allen, J., Alvarez-Jarreta, J., Amode, M. R., Armean, I. M., Austine-Orimoloye, O., Azov, A. G., Barnes, I., Bennett, R., Berry, A., Bhai, J., Bignell, A., Billis, K., Boddu, S., Brooks, L., Charkhchi, M., Cummins, C., Da Rin Fioretto, L., … Flicek, P. (2022). Ensembl 2022. Nucleic Acids Research, 50(D1), D988–D995. https://doi.org/10.1093/nar/gkab1049
Morales, J., Pujar, S., Loveland, J. E., Astashyn, A., Bennett, R., Berry, A., Cox, E., Davidson, C., Ermolaeva, O., Farrell, C. M., Fatima, R., Gil, L., Goldfarb, T., Gonzalez, J. M., Haddad, D., Hardy, M., Hunt, T., Jackson, J., Joardar, V. S., … Murphy, T. D. (2022). A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature, 604(7905), 310–315. https://doi.org/10.1038/s41586-022-04558-8
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., Rajput, B., Robbertse, B., Smith-White, B., Ako-Adjei, D., Astashyn, A., Badretdin, A., Bao, Y., Blinkova, O., Brover, V., Chetvernin, V., Choi, J., Cox, E., Ermolaeva, O., … Pruitt, K. D. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research, 44(D1), D733-45. https://doi.org/10.1093/nar/gkv1189
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 17(5), 405–424. https://doi.org/10.1038/gim.2015.30
Schoch, K., Tan, Q. K.-G., Stong, N., Deak, K. L., McConkie-Rosell, A., McDonald, M. T., Goldstein, D. B., Jiang, Y.-H., & Shashi, V. (2020). Alternative transcripts in variant interpretation: the potential for missed diagnoses and misdiagnoses. Genetics in Medicine : Official Journal of the American College of Medical Genetics, 22(7), 1269–1275. https://doi.org/10.1038/s41436-020-0781-x
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P., & Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature, 456(7221), 470–476. https://doi.org/10.1038/nature07509



