Towards a Future Without Variants of Uncertain Significance (VUS)
Advances in variant classification, computational tools, and collaborative data-sharing improve our diagnostic capabilities to resolve variants of uncertain significance (VUSs); genetic alterations that have unknown effects on patients’ health [1]. These variants impact the decisiveness of genetic tests negatively by causing inconclusive reports [2]. Therefore, resolving all the VUS variants is one of the main goals in the field of human genetics.
In line with this goal, the National Human Genome Research Institute (NHGRI) suggested that all Variants of Uncertain Significance in coding regions of the human genome will be resolved by 2030 in a publication where they made ten ‘‘bold predictions’’ for human genomics by 2030 [3]. Last month, Fowler and Rehm published an opinion piece about the progress made in the last four years to achieve this goal [4]. In this blog piece, we will recap this opinion article, and we will describe how our platform, SEQ, can help the community to resolve VUS cases.
Understanding Variants of Uncertain Significance (VUS) in Human Genetics
The Variant of Uncertain Significance is a genetic alteration identified through genomic testing that lacks sufficient evidence for definitive classification as either benign or pathogenic [1]. Moreover, variants annotated as VUS constitute the largest portion of human genomic variations in which 47.75% of the more than 2 million variants are annotated as ‘Uncertain Significance’ in ClinVar database, a public archive of reports on the relationships among human variations and phenotypes (Figure 1) [5].
Because of the size and ambiguity of these cases, many genetic tests result in inconclusive reports [2]. For example, up to 32% of individuals received inconclusive test reports because of VUS variants from 19 laboratories in North America offering multigene panels, exomes, and genomes in 2020-2021 [6]. Thus, the resolution of these variants will remarkably improve genetic testing outcomes and consequently, patients’ diagnoses.
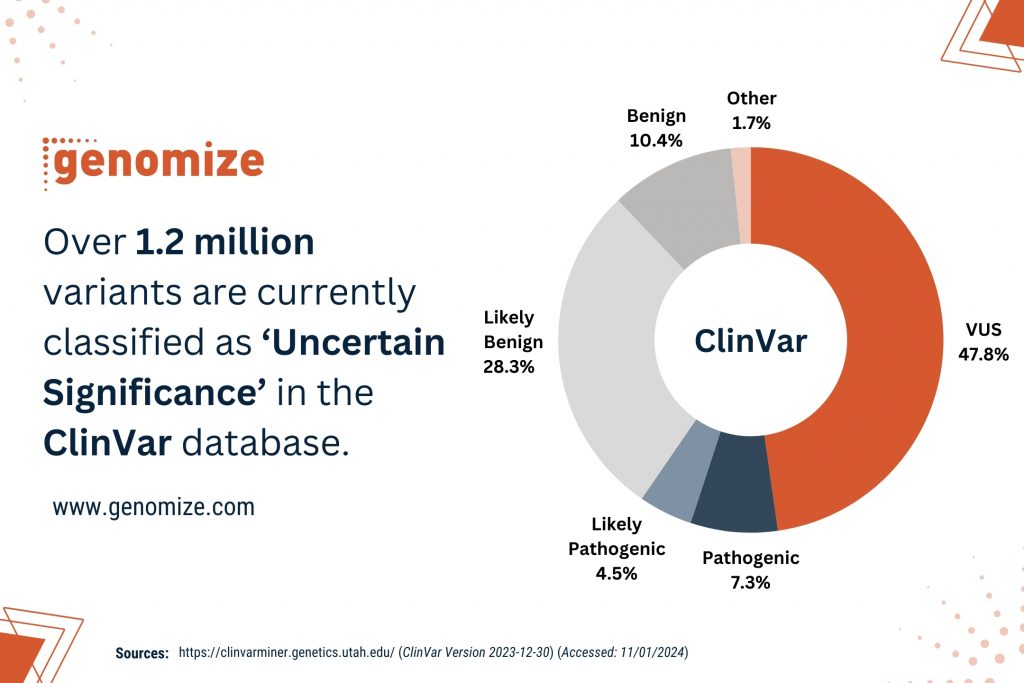
Figure 1: Over 1.2 million variants are currently classified as VUS in the ClinVar database. (Source: https://clinvarminer.genetics.utah.edu/) (Accessed: 11/01/2024)
The Challenge of Resolving VUS: Insights from Fowler and Rehm’s Opinion Piece
In the aforementioned opinion piece [4], Fowler and Rehm emphasize the importance of investing in efforts to eliminate VUS variants, considering their prevalence as one of the major obstacles in precision genomic medicine. They suggest that it is possible to resolve most if not all uncertain significance cases by 2030 thanks to the advances in variant classification standards, enhanced computational tools for predicting variant effects, scalable multiplexed assays offering comprehensive genomic coverage, and collaborative data-sharing initiatives coupled with an increased number of sequenced samples.
On the other hand, they mentioned that the challenges in resolving VUS variants mainly lie in the scarcity of evidence. They claim that the best way to generate the evidence is by performing functional assays for a specific variant. However, they argue that this approach is quite problematic because of the cost and the lack of correct methodology for functional testing. Also, they emphasized that current functional tests mainly focus on single-nucleotide variations, while the functional tests on insertions, deletions, or other structural variants that are important for the disease pathology need further improvements.
Finally, the authors suggest that achieving the goal of eliminating all VUS cases by 2030 is possible but the pace of progress toward this goal depends significantly on the choices that are made in the coming years.
The Role of the SEQ Platform in Resolving VUS Cases
Fowler and Rehm’s opinion piece [4] is one of the most informative articles about the progress in the elimination of most if not all VUS cases by 2030. As Genomize, we believe that the resolution of many exonic VUS cases by 2030 is an achievable target, considering the increase in the accumulated data and the development of more sophisticated computational algorithms such as our platform, SEQ to predict the effect of a variant, which are expected to reduce the amount of VUS cases. With features such as Variant Prioritization [7] and Extended Annotation [8], the SEQ platform is a fast and reliable way to annotate variants to contribute to the resolution of VUS cases.
Fowler and Rehm’s article [4] covers most aspects of our quest to resolve all VUS variants, except an important point that needs to be further examined. That point is the fact that although the number of sequenced samples is increasing, this increase unfortunately is not uniform. For example, after the last release, the Genome Aggregation Database (gnomAD v4) now consists of 807,162 samples, of which 77.07% of these samples are from people with European Ancestry [10]. Similarly, other genomic databases such as 1000 Genomes and ESP are also skewed towards populations with European Ancestry [11].
Moreover, VUS variants are most commonly reported among underrepresented populations because of the scarcity of data and genomics projects [12,13]. Therefore, variants that are present in populations that are non-represented or underrepresented in databases might create a problem for the goal of resolving VUS cases. This is because a higher frequency of VUS variants in these populations could unravel the pathogenicity of these variants and remove possible biases in their prioritization. Therefore, solutions to accumulate data from these populations would be crucial in resolving all VUS cases.
As Genomize, we noticed this problem and developed “the data aggregation function” in our software, the SEQ Platform. By using our platform, our users can establish their real-time phenotype-genotype database (a center-specific genome project in essence) to generate the frequency of each variant in their samples. This could be very important because by using this frequency data, each variant can be evaluated separately and common variants in a population can be easily annotated for further analyses. We also believe that this function will significantly improve the resolution of VUS variants and help the community to achieve the goal of resolving all exonic variants by 2030.
References
1. Richards S, et al. (2015). “Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology”. guideline. Genetics in Medicine. 17 (5): 405–24.
2. Vears DF, et al. (2017) “Reporting practices for variants of uncertain significance from next-generation sequencing technologies”. Eur J Med Genet. 60(10):553-558. doi: 10.1016/j.ejmg.2017.07.016. PMID: 28774848.
3. Green, E.D., et al. (2020) “Strategic vision for improving human health” at The Forefront of Genomics. Nature 586, 683–692. https://doi.org/10.1038/s41586-020-2817-4.
4. Fowler DM and Rehm HL. (2023) “Will variants of uncertain significance still exist in 2030?” Am J Hum Genet. S0002-9297(23)00400-7. doi: 10.1016/j.ajhg.2023.11.005. Epub ahead of print. PMID: 38086381.
5. https://clinvarminer.genetics.utah.edu/variants-by-significance (Accessed: 11/01/2024)
6. Rehm, H.L., et al. (2023). “The landscape of reported VUS in multi-gene panel and genomic testing: Time for a change”. Genet. Med. 25, 100947.
7. https://genomize.com/ai-driven-ngs-data-analysis-a-variant-prioritization-pipeline-for-precision-medicine (Accessed: 11/01/2024)
8. https://genomize.com/extended-annotation-solution-for-ngs-data-analysis (Accessed: 11/01/2024)
9. https://genomize.com/about_us (Accessed: 11/01/2024)
10. https://gnomad.broadinstitute.org/news/2023-11-gnomad-v4-0 (Accessed: 11/01/2024)
11. Marwaha S, Knowles JW, Ashley EA. (2022) “A guide for the diagnosis of rare and undiagnosed disease: beyond the exome”. Genome Med. 14(1):23. doi: 10.1186/s13073-022-01026-w. PMID: 35220969; PMCID: PMC8883622.
12. Caswell-Jin JL, et al. (2018). “Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk”. Genet Med. 20(2):234-239. doi: 10.1038/gim.2017.96. Epub 2017 Jul 27. PMID: 28749474.
13. Chen E, et al. (2023). “Rates and Classification of Variants of Uncertain Significance in Hereditary Disease Genetic Testing”. JAMA Netw Open. 6(10):e2339571. doi:10.1001/jamanetworkopen.2023.39571. PMID: 37878314; PMCID: PMC10600581.